Package Exports
This package does not declare an exports field, so the exports above have been automatically detected and optimized by JSPM instead. If any package subpath is missing, it is recommended to post an issue to the original package (seqera) to support the "exports" field. If that is not possible, create a JSPM override to customize the exports field for this package.
Readme
Seqera AI CLI
AI-powered assistant for bioinformatics workflows and Seqera Platform.
Beta: Seqera AI CLI is currently in beta. Features and commands may change as we continue to improve the product.
Note: Seqera Cloud users receive $20 in free credits to get started with Seqera AI. Contact us for additional credits.
Get started
Install the Seqera AI CLI:
pip install seqeraSee Installation for a comprehensive installation guide.
Log in to your Seqera account:
seqera login
See Authentication for a comprehensive authentication guide.
Start Seqera AI:
seqera ai
Run your first prompt:
/debugSee Use cases for a comprehensive list of use cases.
Use cases
Seqera AI is an intelligent command-line assistant that helps you build, run, and manage bioinformatics workflows. The following sections describe several common use cases.
Work with Nextflow
Seqera AI helps you develop, debug, and understand Nextflow pipelines with AI-powered analysis and code generation.
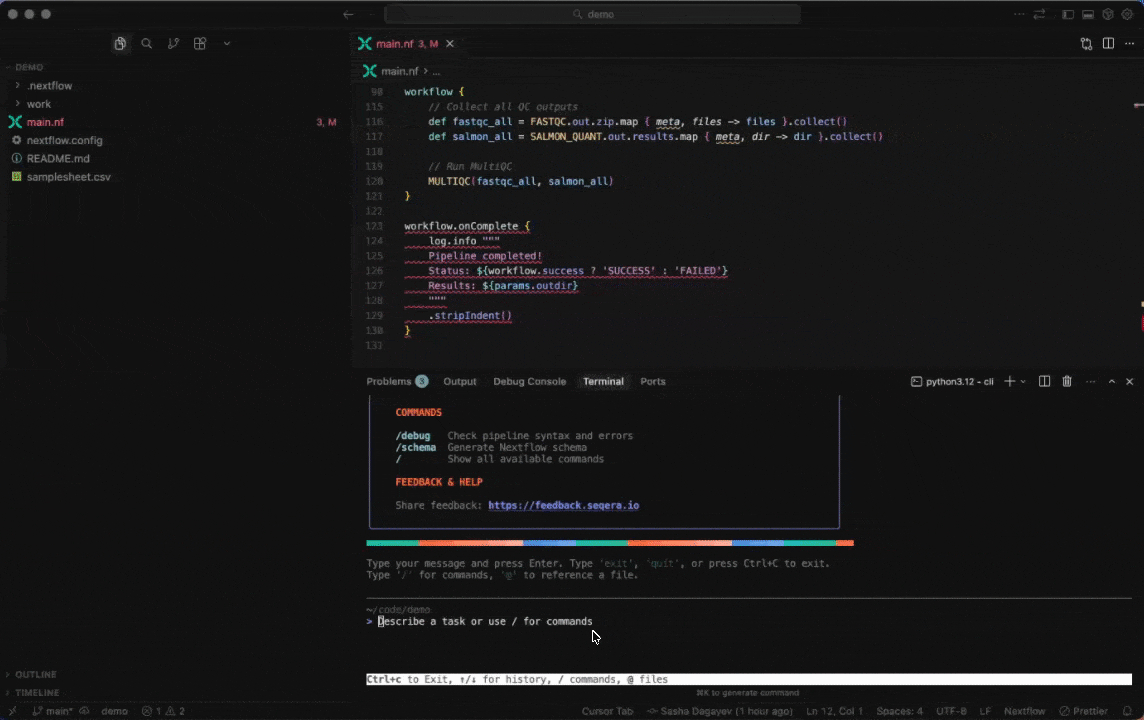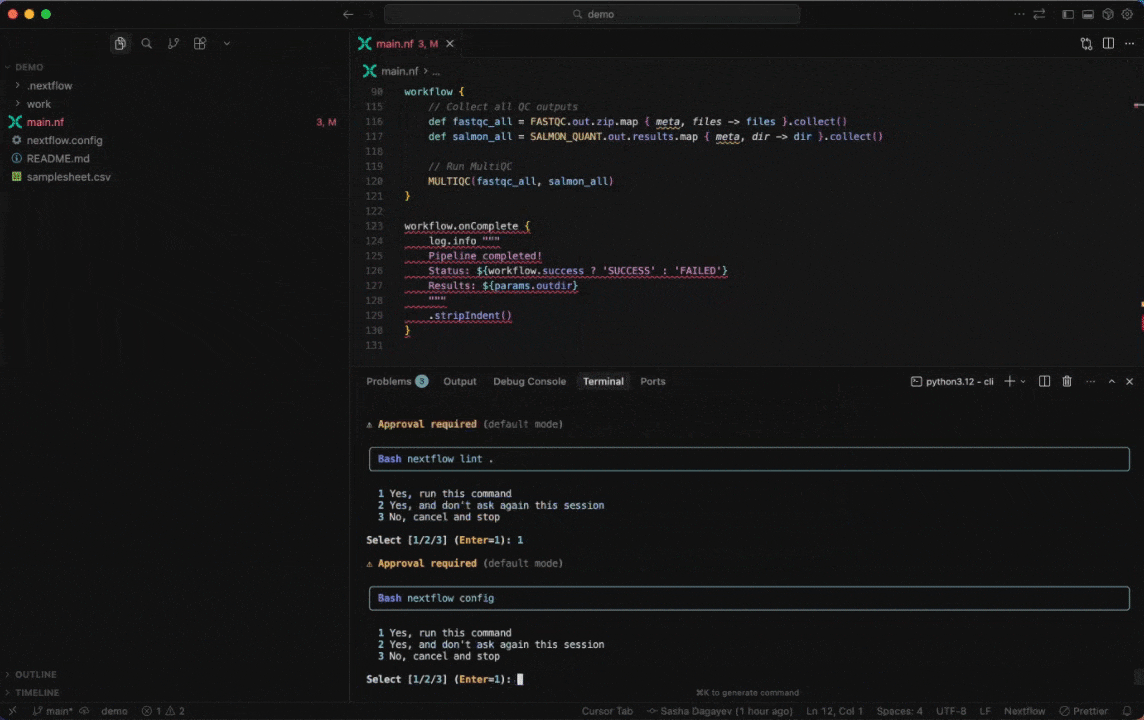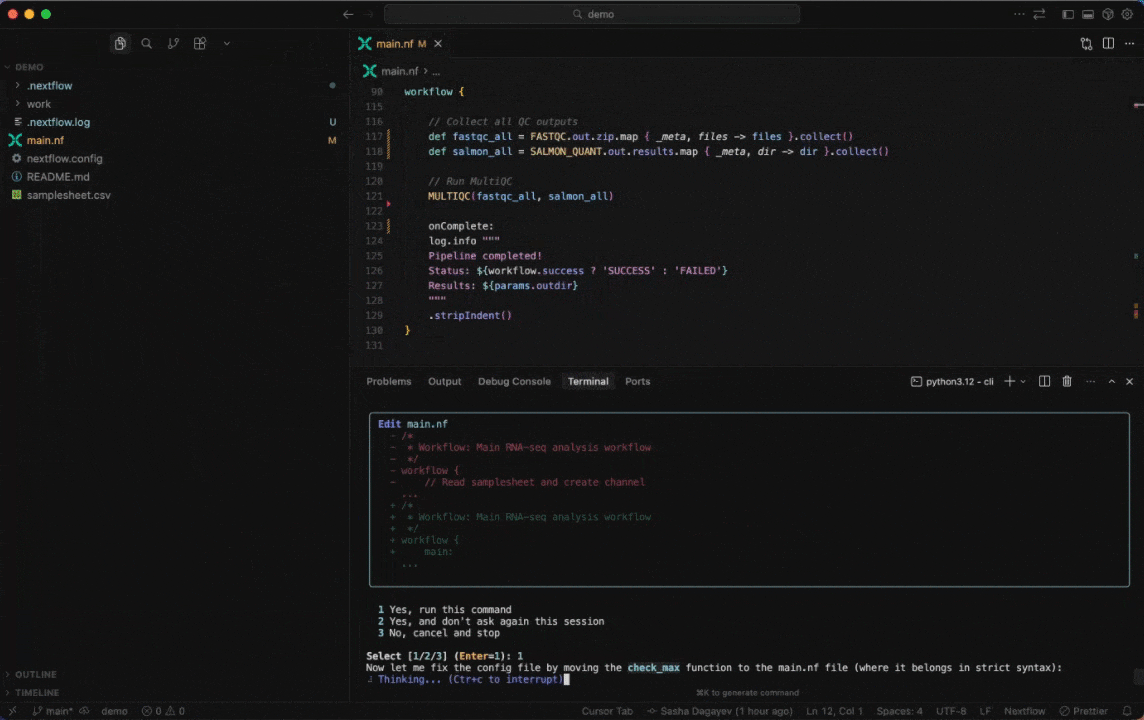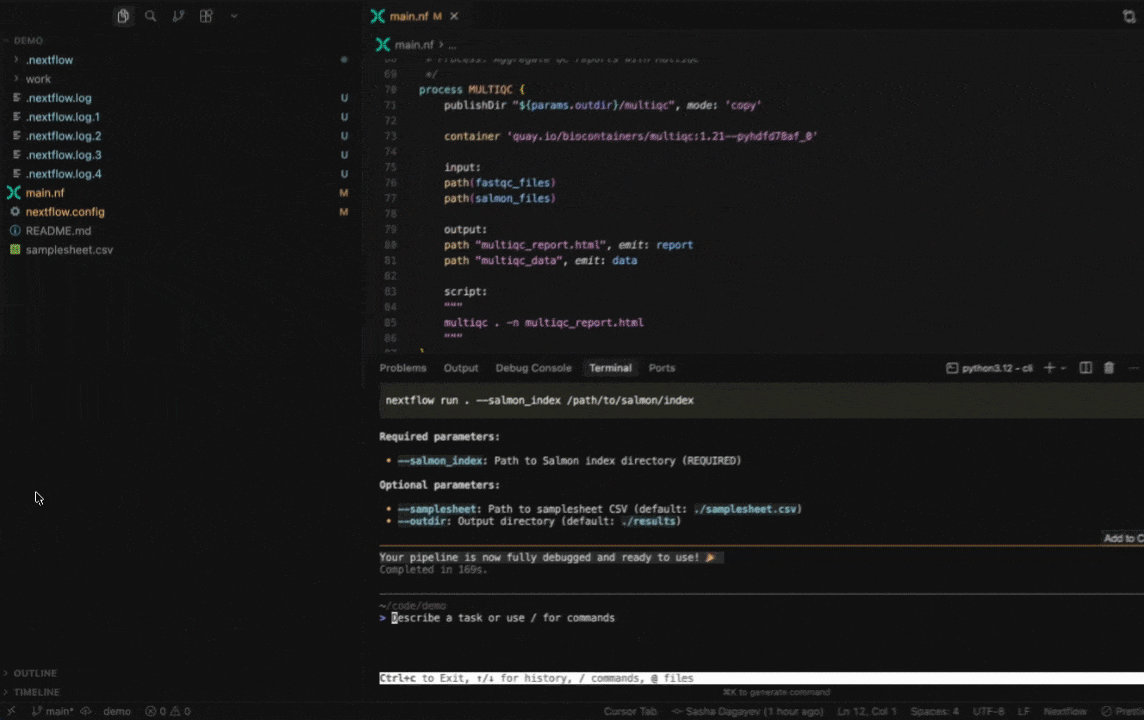
Working with Nextflow
Understand your pipeline structure:
> Show me the structure of main.nf> What processes are defined in this pipeline?Generate a nextflow.config file:
> /configDebug your pipeline:
> /debug> Why is my pipeline failing?Generate a schema (nextflow_schema.json) file:
> /schemaConvert scripts to Nextflow:
> /convert-python-scriptBuild containers with Wave
Seqera AI can create containerized environments using Wave, without requiring you to write Dockerfiles.
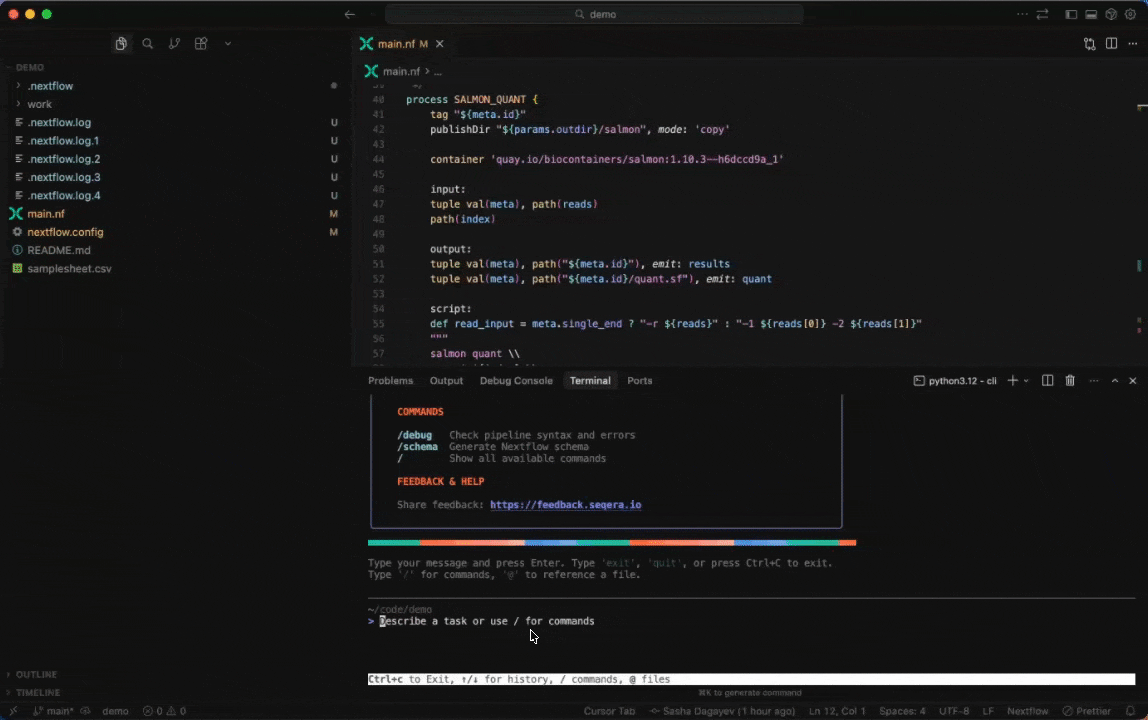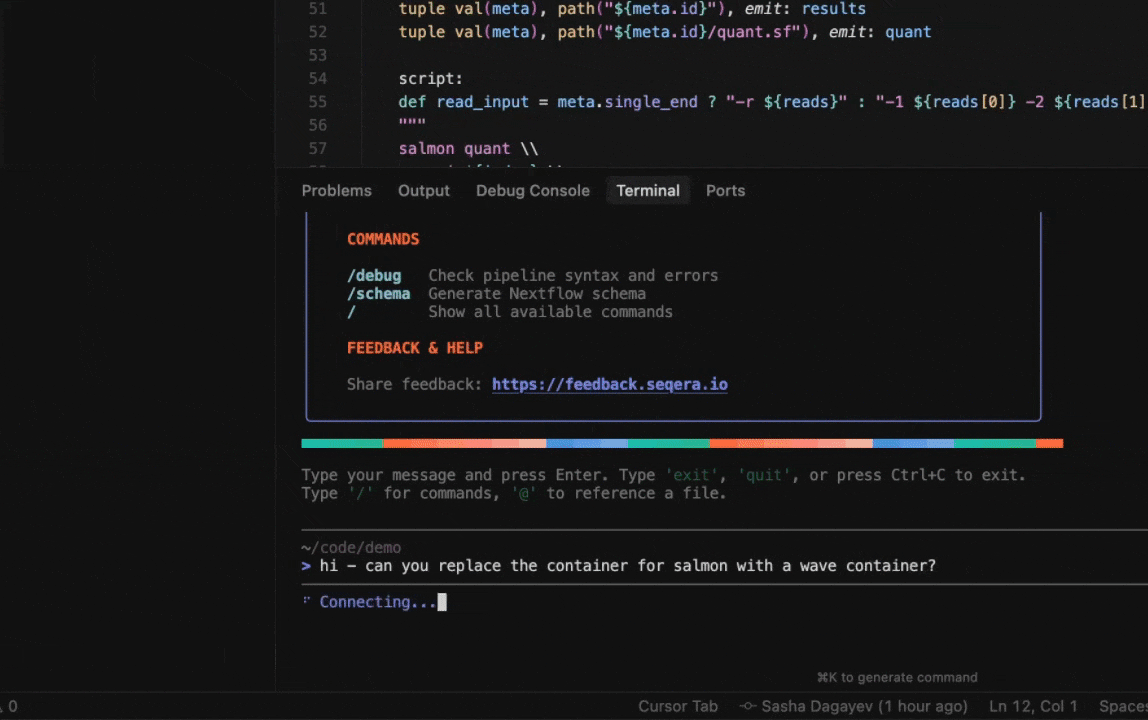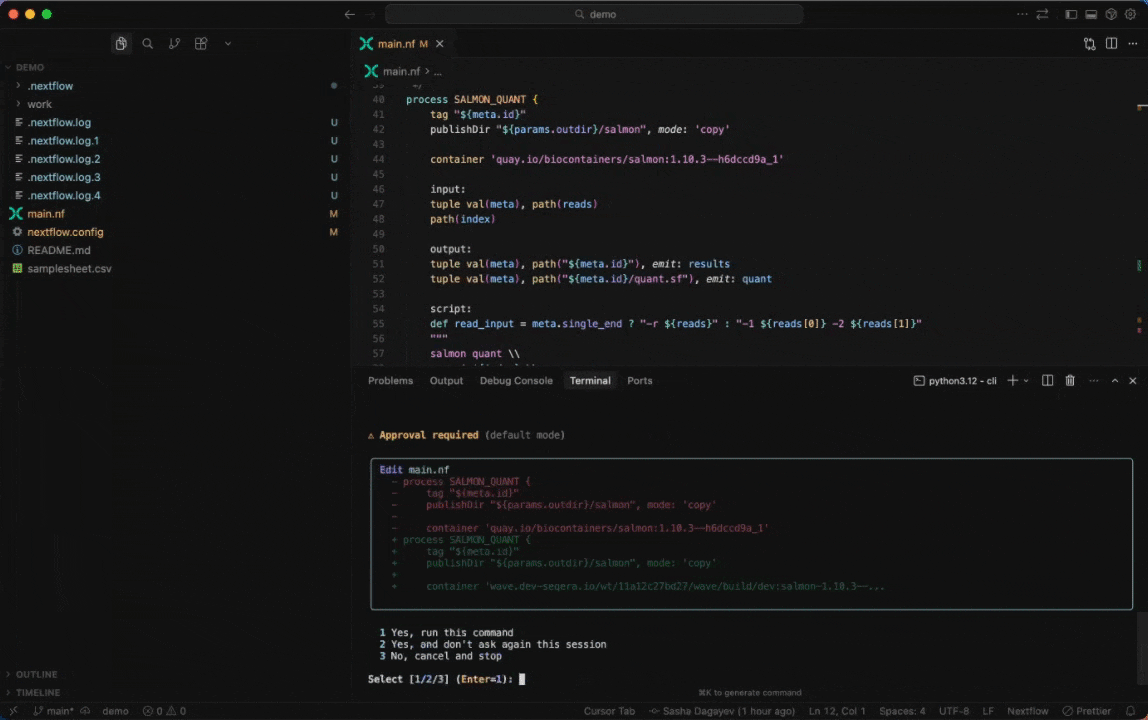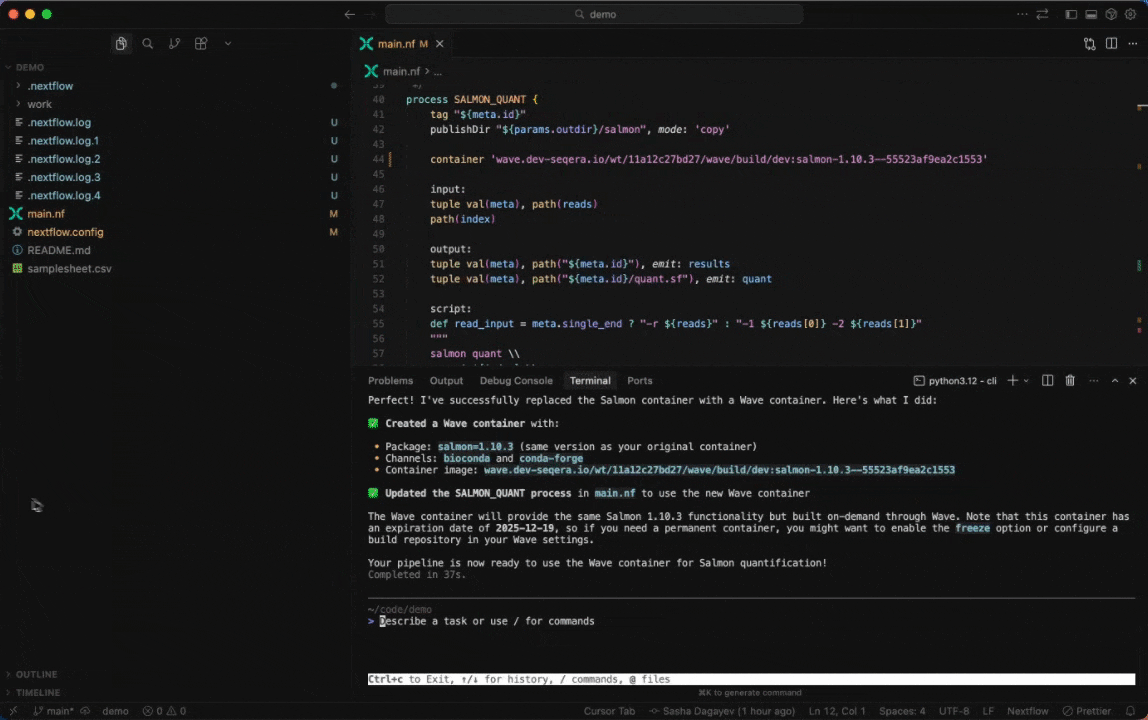
Building containers with Wave
Create a container with conda packages:
> Create a container with samtools and bwa from biocondaCreate a container with pip packages:
> Build a container with pandas, numpy, and scikit-learnGet a container for a specific tool:
> I need a container with FastQC version 0.12.1Note: The assistant will generate a Wave container URL that you can use directly in your Nextflow pipelines or pull with Docker.
Customize your session
Customize your session with command-line options.
Customize your session
Start in a specific directory:
seqera ai -w /path/to/projectSet approval mode for local commands:
seqera ai -a fullExit the assistant
End your Seqera AI session when done.
Exit the assistant
To end your session:
- Type
exitorquit - Press
Ctrl+C
Note: Your conversation history is preserved for the session but not stored permanently.
Use slash commands
Seqera AI includes built-in slash commands for common workflows.
Use slash commands
Type / to see all available commands:
| Command | Description |
|---|---|
/config |
Generate a nextflow.config file |
/schema |
Generate a Nextflow schema |
/debug |
Run nextflow lint and preview |
/debug-last-run |
Debug the last local run |
/debug-last-run-on-seqera |
Debug the last Platform run |
/migrate-from-wdl |
Convert WDL to Nextflow |
/migrate-from-snakemake |
Convert Snakemake to Nextflow |
/convert-python-script |
Convert Python script to Nextflow |
/convert-r-script |
Convert R script to Nextflow |
/convert-jupyter-notebook |
Convert Jupyter notebook to Nextflow |
/write-nf-test |
Write nf-tests for your pipeline |
Work with data
Seqera AI helps you manage data through Platform data links and access reference datasets.
Working with data
Browse data links:
> List my data links> Show me the contents of my S3 data linkDownload and upload files:
> Generate a download URL for results/final_report.html> Upload my local results to the data linkAccess reference data:
> Find the human reference genome GRCh38> Search for RNA-Seq test dataWork with local files
Seqera AI can interact with files in your current working directory.
Work with local files
Start the assistant from your project folder:
cd /path/to/your/project
seqera aiThen, ask the assistant to help with local tasks:
> Show me the structure of main.nf> Add a new process to handle quality controlNote: Local file operations are controlled by approval modes. By default, the assistant will ask for your approval before making changes outside your working directory or running potentially dangerous commands.
Work with nf-core modules
Seqera AI provides access to over 1,000 nf-core modules for common bioinformatics tasks.
Working with nf-core modules
Search for modules:
> Find nf-core modules for sequence alignment> What modules are available for variant calling?Get module details:
> Show me how to use the nf-core/bwa/mem moduleRun a module:
> Run FastQC on my FASTQ filesNote: The assistant can generate the exact Nextflow command with proper parameters for your data.
Work with Seqera Platform
Use Seqera Platform capabilities to run and manage workflows at scale with AI assistance.
Working with Seqera Platform
List your workflows:
> List my recent workflowsLaunch a pipeline:
> Launch the nf-core/rnaseq pipeline with the test profileDebug failed runs:
> Why did my last workflow fail?> Get the logs for the failed task in my last runLearn more
- Installation: Detailed installation instructions
- Authentication: Log in, log out, and session management
- Command approval: Control which commands run automatically
- Use cases: Seqera AI CLI use cases
- Troubleshooting: Troubleshoot common errors